TRENDNMR
TREND NMR – Binding Isotherms and Time Courses Readily from NMR
- TREND NMR is an NMR-optimized improvement upon Trendmain. It requires a minimum of five measurements (collected uniformly) in order to track a trend among them or to make a comparison.
- In the left side bar, there are three modes to choose from:
spectra, fid, and peaklist. They function similarly, but differ in small ways.
- For details of each option, see the GUI manual and CLI manual).
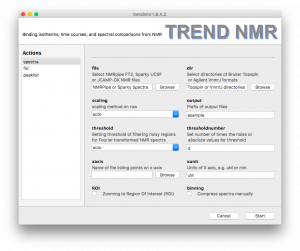
- Spectra
- The
spectra menu is used for analysis of NMR spectra. It is similar to the nmr menu of Trendmain.
TREND NMR automatically selects the format of NMR spectra. Under the file heading, there is a file browsing function for choosing a series of 2D NMR spectra in NMRPipe ft2, Sparky ucsf, or jcamp-dx formats.- Under the
dir heading, there is a directory browsing function for choosing a series of directories containing hFourier-transformed Bruker Topspin spectra (brukerspectra)(by experiment number) or Agilent VnmrJ (agilentspectra)
- Scaling is important for an accurate binding isotherm. Usually
auto is the best choice, but sometimes pareto is more accurate. The default scaling method of rows of the data matrix is auto, which is recommended for series of NMR spectra in fast or slow exchange regimes. pareto is recommended for spectra with intermediate exchange behavior. Considerations in choosing scaling method and setting the threshold to filter out noise are given in the Supporting Information of Jia Xu and Steven R. Van Doren, Binding Isotherms and Time Courses Readily from Magnetic Resonance. Anal. Chem. 2016, 88 (16), pp 8172-8178
threshold and thresholdnumber can be set for filtering noise out of nMR spectra. The three ways to set threshold are auto, absolute, and the number of times the noise level. In auto mode, the program determines noise from the first spectrum and set the threshold as 4-fold the noise level for autoscaling and 0.5 times the noise level of Pareto scaling.Compression of the spectra can be turned on for a faster, more economical calculation, if desired. The number of points to group together can be adjusted interactively using a slider.- The
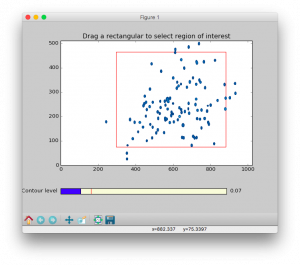
ROI option allows zooming in on the region of interest (ROI). This is useful for avoiding disruptive spectral artefacts, e.g. from the water or other solvent. When the ROI option is turned on, a preview of the first spectrum in the series will pop up. The ROI can be selected by dragging a rectangular of the spectrum using mouse. After this window is closed, a preview of selected ROI will pop up and be saved as a PNG picture with suffix of -ROI.png to the directory of input spectra.
- The contour level can be adjusted by dragging the
Contour level widget in the bottom of the preview.
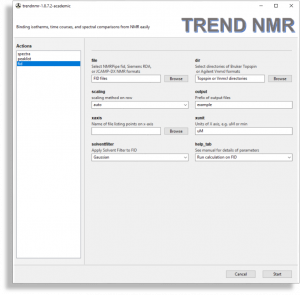
- FIDs
- The
fid menu is very similar to the spectra menu but should be used if analyzing free induction decays (FIDs). This menu is simplified for handling FIDs, but adds the solventfilter option for subtrcting an on-resonance solvent signal. FID files such as NMRPipe fid, Simens RDA, JCAMP-DX formats are supported by the file browser. Directories each containing a Bruker Topspin FID (brukerfid) or Agilent VnmrJ FID (agilentfid) can be read by the dir browser.
- Peaklists
- TREND NMR reads peak lists in
Sparky and NMR-STAR formats in its peaklist mode. Multiple pieces of NMR software can export peak lists in Sparky and NMR-STAR formats. TREND NMR can retrieve NMR-STAR files archieved at the Biological Magnetic Resonance Data Bank (BMRB) when the accession codes are listed in a text file.
It supports the scaling of rows specified in the scaling field. There is also the option to scale columns, which is specified in the columnscaling field. trendmaingui (the program from the original, general-purpose TREND package) also reads and analyzes lists in Excel, CSV, and text files, any of which would be suitable for peak lists.
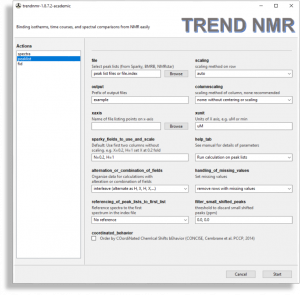
- Please configure this if not using 15N HSQC or TROSY spectra: Under
sparky_fields_to_use_and_scale, the default of “N=0.2, H=1” is appropriate for selecting and scaling the correct fields from amide peak lists. (These peak lists are generated from widely used 15N HSQC and TROSY NMR spectra.) For peak lists from 13C-1H correlation spectra such as heteronuclear single quantum coherence (HSQC), heteronuclear multiple quantum coherence (HMQC), and methyl TROSY, a similar choice of “C=0.25, H=1” is suitable. Peak lists from 2D homonuclear 1H-1H total correlation spectroscopy (TOCSY) or nuclear Overhauser effect and exchange spectroscopy (NOESY) could instead be set to “H=1, H=1”.
handling_of_missing_values is used to process NMR peak lists differing in the number of peaks. The default of remove rows with missing values deletes from the calculation those peaks not present in all of the lists. use mean among spectra handles missing peaks by filling the mean value from the other spectra. use mean within spectrum sets missing values as the mean of the other peaks in the spectrum. set to zero fills in missing values with 0 for the calculation.alternation_or_combination_of_fields organizes chemical shift data for the calculation. The default option of interleave (alternate as H, X, H, X, ....) option reorganizes scaled chemical shift data from each spectrum (scaled by field sparky_fields_to_use_and_scale) into a 1D vector for PCA or CONCISE calculation. See Sakurai et al PNAS 2007 104 (39) 15346-1535.
The combine (H+X, H+X, ...) option reduces the dimensionality of 2D or multi-dimensional spectra by calculating combined chemical shfit (CCS) use the weighting factor defined in sparky_fields_to_use_and_scale. For example, the CCS of HSQC (“N=0.2, H=1”) is calculated as CCS =0.2×N+1×H. See Boulton et al, Sci Rep, 2014 Dec 8;4:7306.referencing_of_peak_lists_to_first_list This can be used to reference spectra to the first spectrum in the index file for PCA or CONCISE calculation.
By default, this option is turned off because it assumes the input peak lists are already referenced. The Linear style of referencing is recommended for CONCISE. (See p6511 in Cembran et all PCCP 2014). Referencing using root mean squared (RMS) differences (see Xu et al, Sci Rep, 2016) between peak positions may also be chosen but can decrease the number of peaks retained by CONCISE.
can decrease the number of peaks retained by CONCISE.filter_small_shifted_peaks sets the threshold (in ppm) to discard peaks that shift too little. The threshold values are separated by commas in the field called sparky_fields_to_use_and_scale. The field is by default turned off (0.0, 0.0). When sparky_fields_to_use_and_scale is set as default N=0.2, H=1 and alternation_or_combination_of_fields is set to the default of interleave, a 0.02, 0.02 setting of filter_small_shifted_peaks discard rows (either N or H) whose values vary less than 0.02 ppm across the spectra (i.e. columns). If the alternation_or_combination_of_fields is set to ‘combine’, the first values in filter_small_shifted_peaks is used to filter peaks whose CCS vary less than the threshold (e.g. 0.02 ppm) across different spectra.coordinated_behavior This checkbox is checked to turn on CONCISE analysis when start button is clicked at the lower right to start the calculation. TRENDanalysis can be used to continue and adjust the calculations under the CONCISE tab. For example, the first and last extreme states may be set. The plot can be labeled using a file listing the labels for the peaks. See the CONCISE manual for details.